It can be defined as “Changing the genes of an organism by using in vitro processes.”
The first recombinant DNA was constructed by Stanley Cohen and Harbert Boyer (1973). They cut the piece of DNA from a plasmid carrying antibiotic resistance gene in the bacterium Salmonella typhimyreum and linked it to plasmid of Escherichia coli. This rDNA was introduced into E. coli and made to multiply (gene cloning) making a number of replicas.
Strategies of Recombinant DNA technology:
It involves the following steps-
Isolation of a DNA fragment containing a gene of interest that needs to be cloned (called as insert).
Generation of a rDNA molecule by insertion of the DNA fragment into carrier DNA molecule called vector (for example Plasmid) that can self-replicate within the host cells.
Transfer of the rDNA into a host cell (for example E.coli).
Identification of transformed cells (i.e. cells carrying rDNA) and their selection from non-transformants.
Amplification of rDNA to get its multiple copies in a cell.
Cell multiplication to get a clone this facilitates each of clones to possess multiple copies of foreign DNA.
Basic tools of Recombinant DNA technology:
The key tools needed for the rDNA technology to be accomplished are:
Cell culture with desired DNA
Restriction enzyme
DNA polymerase
Ligase
Vector
Host organism/cell
Restriction Enzyme:
The restriction enzyme are called as molecular scissors. These act as foundation of rDNA technology.
Restriction enzymes are present in bacteria and provide a type of defense mechanism called the restriction –modification system.
Molecular basis of these systems was elucidated first by Werner Arber in 1965.
This system consists of two components-
A restriction enzyme that selectively recognize a specific DNA sequence and digest any DNA fragment containing that sequence.
A modification enzyme that adds a methyl groups to one or two bases within the sequence recognized by the enzyme.
Types of Restriction enzyme:
There are three main type of restriction endonucleases. These are designated as Type I, Type II, and Type III. Type I: this enzyme cuts DNA at random sites that can be more than 1000 base pairs from the recognition sequence. These move along the DNA in a reaction and require MG++, S-adenosyl methionine ATP as co-factor. Type II: this type of restriction enzyme was first isolated by Hamilton Smith. These are simple and require no ATP for degradation of DNA. They cut DNA within the recognition sequence. Type III: this group of endonucleases cut the DNA about 25 base pairs from the recognition sequence. In a reaction, it moves along the DNA and requires ATP as source of energy.
Naming of restriction enzyme:
The first letter of the name comes from the genus and the next two letters from the name of the species of the prokaryotic cell from which they are isolated.
The next letter comes from the strain of the prokaryote.
The roman numbers following these four letters indicates the order in which the enzymes were are isolated from the strain of bacterium. For example-
ECOR I is isolated from Escherichia coli, RY 13.
Hind II is isolated from Haemophilus influenza.
Bam H I is from Bacillus amyloliquefaciens.
Restriction enzyme were first discovered and studied by the molecular biologists W. Arber, H. Smith and D. Nathans for which they were awarded the Nobel Prize in 1978.
Cleavage of recognition sequence:
Type II restriction enzymes are used in gene manipulation because they can be used in vitro to cut the DNA within the specific sequence typically consisting of 4 to 8 base pairs. This sequence is referred to as Restriction site (Recognition sequence) and is generally palindromic which means that the sequence in both DNA strand at this site read same 5ing specific palindromic sequence. The 5’ to 3’ direction.
For example: ECOR I binds to a region of having specific palindromic sequence. The length of this sequence is 6 base pairs. It cut between ‘G’ and ‘A’ residues of each span and produces two single stranded complimentary cut ends, which are asymmetrical. These ends are called sticky ends or cohesive ends. Because nucleotide bases of this region can pair and stick the DNA fragment again.
Some enzymes like Alu I cleave both strand of DNA through the centre resulting in blunt or flush end. These are also known as symmetrical cuts.
Restriction fragment length polymorphism (RFLP):
DNA of each organism has specific sequence which can be cleaved by several restriction enzymes and fragment of different length can be produced. These fragments are called restriction fragments.
Restriction fragments of different individuals and species varies, these are of different length because of variation in DNA sequence of restriction sites. These variations are referred to as Restriction fragment length polymorphism (RFLP).
Uses of othef enzumes in Recombinant DNA technology
There are many other enzymes too which are used in rDNA technology. Some of the important enzymes are given below: DNA Polymerase I:
It is used in synthesis of DNA complementary to DNA template in 5’ to 3’ direction. It lacks 5’ to 3’ exocatalytic activity. Exonuclease III:
It cleaves from the end of a linear DNA and digests double stranded DNA from the 3’ end only. DNA Ligase:
DNA Ligase has the physiological role in stealing single strand necks in the double stranded DNA. It forms phosphodiester bond between two adjacent nucleotides. The reaction requires one of the fragments to have a 5’ phosphate residue and the other have a 3’ hydroxyl group.
There are two types of DNA ligase. E. coli DNA ligase (which used NAD as source of energy) and T4 DNA ligase (which used ATP as source of energy). Alkaline phosphate (AP):
The 5’ phosphate group is essentially required for ligation of DNA fragments, but DNA cannot be ligated if 5’ phosphate group is removed. The enzyme is used to check self ligaion of vector DNA where there is a problem of recircularisation. The sources of alkaline phosphate are bacteria (BAP) and calf intestine (CAP).
Cosmid Vectors:
Cosmid have been constructed by combining certain features of plasmid and ‘cos’ site of phage λ (lamda).
Cosmid vector possesses an ‘ori’ (origin of replication) gene, a selectable genetic marker (for antibiotic resistance), suitable recognition sites for restriction enzymes (i.e. cloning site) and cohesive cos site.
Cosmid can be used to clone DNA fragments up to 45Kbp in size.
Yac Vectors:
YACs or Yeast Artificial Chromosomes are used as vectors to clone DNA fragments of more than 1 Mb in size.
It has been used for physical mapping of human chromosome in ‘Human Genome Project’.
The YAC vector contains the E.coli origin of replication (oriE) and a selectable marker (ampr), a yeast DNA sequence, centromere pathway (TRP) and telomeric (T) sequence.
There are recognition sites for restriction enzymes such as Sma I and Bam HI.
Bac Vectors:
BACs are Bacterial Artificial Chromosomes are vectors based on the natural extra chromosomal plasmid from E.coli, the fertility or F plasmid.
BAC vectors contain ori gene, a gene for maintainence of F factor a selectable marker (an antibiotic resistance gene) and many restriction sites for insertion of foreign DNA.
These vectors can accommodate inserts up to 500 Kb and are used in genome sequencing projects.
Animal and Plant Viral Vectors:
Viruses that infect plant and animal cells have also been manipulated to introduce foreign genes into plant and animal cells.
The natural ability of viruses to adsorb to cells, introduce their DNA and replicate, have made them ideal vehicle to transfer foreign DNA into eukaryotic cells in culture.
Animal Viral Vectors:
A vector based on Simian virus 40 (SV 40) was used in the first cloning experiment involving mammalian cells.
At present, retroviral vectors are popular for cloning genes in mammalian cells.
Plant Viral Vectors:
In case of plants, viruses like Cauliflower Mosaic virus (CaMV), Tobacco Mosaic Virus (TMV) and Gemini Virus (GV) have been used with limited success.
Host Cells
For the multiplication of foreign DNA efficient host cells are required.
Different types of host cells such as E.coli, Yeast, plant and animal cells are available for gene cloning. These cells are used according to the aim of experiment.
E.coli has become the most widely used organism in rDNA technology because
Its genetic makeup has been extensively studied.
It is easy to handle and grow, can accept a range of vectors and has been extensively studied for safety.
It doubles its cell number in each 20 mins.
Within hour thousands of bacteria cells and that much number of foreign genes are produced and its recombinant proteins are expressed.
For expression of eukaryotic gene, the eukaryotic hosts like Yeast cells are exploited, like E.coli, Yeast also has several advantages. Such as
Easy to grow and manipulate.
Simplest unicellular eukaryotes.
Well characterized.
Requirement of no complex medium.
Their multiplication in laboratory in a small vessel or a large sized container.
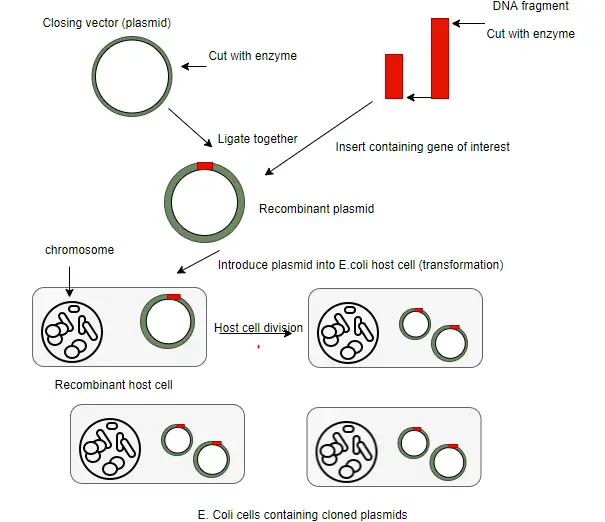
Construction of Recombinant DNA(rDNA)
The first step in the construction of a rDNA molecule is to isolate the vector and the fragments containing the gene to be cloned.
The vector and target DNA fragments are separately digested with the same restriction enzyme such as ECOR I which generate sticky ends.
The vector is then treated with alkaline phosphatase enzyme so that later in the ligation step the vector does not self ligate.
The cut vector and DNA fragments are mixed in a suitable ratio and ligated with the enzyme DNA ligase to yield a recombinant vector containing insert.
Introduction of rDNA into host cells
The recombinant vector carrying foreign DNA needs to be transferred into the suitable host cell. These are several methods to introduce recombinant vectors and these are dependent on several factors such as the vector type and host cell.
Some commonly used procedures are as follows:
Transformation
In rDNA technology the most common method to introduce rDNA into living cell are called transformation.
However, in nature the frequency of transformation of many cells (example yeast and mammalian cells) is very less. Secondly, all the time host cells do not undergo transformation, because they are not prepared for it.
When they develop competence factors in the cells, transformation phenomenon occurs.
There are some factors which affect transformation such as concentration of foreign DNA molecule, host’s cell density, temperature etc.
In this procedure, bacterial cells take up DNA from the surrounding environment.
In 1970, Mendel and Hugo found that E.coli cells become markabely competent to take up external DNA when suspended briefly in cold calcium chloride (CaCl2) solution.
Transfection
Transfection is the transfer of foreign DNA into cultured host cells mediated through chemicals.
The charged chemical substances such as cationic liposome, calcium phosphate of DEAE dextran are taken and mixed with DNA molecules.
The recipient host cells are overlayed by this mixture.
Electroporation
An electric current is used to create transient microscopic pores in the recipient host cell membrane allowing recombinant DNA to enter.
Microunjection
Exogenous DNA can also be introduced directly into animal and plant cells without the use of eukaryotic vectors.
In the procedure of microinjection, foreign DNA is directly injected into recipient cells using a fine micro syringe under a phase contrast microscope to aid vision.
Biolistics
Microscopic particles of gold or tungsten are coated with the DNA of interest and bombarded onto cells with a device much like a particle gun. Hence the term biolistics is used.
Note: Another method of introducing foreign genes is by natural genetic engineer Agrobacterium tumefaciens.
Identification of Recombinants
After the introduction of rDNA into suitable host cells, it is essential to identify those cells which have received the rDNA molecules. This process is called screening or selection.
Generally, the selection methods are based on the expression or non-expression of certain traits such as antibiotic resistance, expression of an enzyme such as β- galactosidase or protein such as GFP (Green Fluorescent Protein) and dependence or independence of a nutritional requirement such as the amino acid leucine. Direct Selection of Recombinants
If the host E.coli cells have taken up the plasmid pBR322, then these cells will grow in media containing the antibiotic ampicillin or tetracycline whereas normal E.coli cells will be killed by the antibiotics. Thus, only transformed cells, however few, will be selected for growth and division. Insertional Selection inactivation Method
This is more efficient method than the direct selection. In this approach, one of the genetic trait is disrupted by inserting foreign DNA.
Antibiotic resistance genes act as a good insertion inactivation system.
Plasmid pBR322 contains two antibiotic resistance genes, one for ampicillin (ampr gene), and the other for tetracycline (tetr gene). If the target DNA is inserted into tetr gene using Bam HI, the property of resistance to tetracycline will be lost. Such recombinants would be test sensitive.
When such recombinants (containing target DNA in tetr gene) are grown into medium containing tetracycline, they will not grow because their tetr gene has been inactivated. But they are resistant to ampicillin because ampr gene is functional.
On the other hand, the self-ligated recombinants will show resistance to ampicillin and tetracycline. Therefore, they will grow on medium containing both the antibiotics. blue- white selection Method
Another powerful method of screening for the presence of recombinant plasmid is referred to as blue-white selection.
This method is based upon the insertional inactivation of the lac-Z gene present on the vector (e.g. PUC19).
This gene expresses the enzyme β- galactosidase whose activity can cleave a colourless substrate called X-gal into blue colored product.
If the lac-Z gene is inactivated due to the presence of the insert, then the enzymes is not expresses. Hence, if after a transformation experiment the E.coli host cells are plated on an ampicillin and X-gal containing solid media plate then colonies which appears blue are those which have transformed cells (antibiotic resistant) but do not have the insert (express active enzyme).
Colonies which appear white are both ampicillin resistant and have the insert rDNA and thus are the cells to be used for future experiment.
Polymerase Chain Reaction (PCR)
In 1983, Kary Mullis invented the polymerase chain reaction (PCR).
The basic principle under laying this technique is that when a double stranded DNA molecule is heated to a high temperature, the two DNA strands separate giving rise to single stranded DNA molecules which can be made to hybridize with small oligonucleotide primer (single stranded) by bringing down the temperature. If to this an enzyme called DNA polymerase and nucleotide tri phosphate is added, much like what happen during replication i.e. primer extension occurs.
This event is repeated several times; therefore, original DNA strand is produced in multiple copies. Due to reaction in several cycle, it is called polymerase chain reaction (PCR).
The basic requirements of PCR reactions are:
DNA template to be amplified.
Primers which are oligonucleotides, usually 10-18 nucleotides long, that hybridize to the target DNA, one to each strand of DNA.
DNA polymerase enzyme which is stable at temperature above 80°C. Taq polymerase which has been isolated from a thermostable bacterial species, Thermus aquaticus, is used.
Deoxynucleotide triphosphate and PCR buffer.
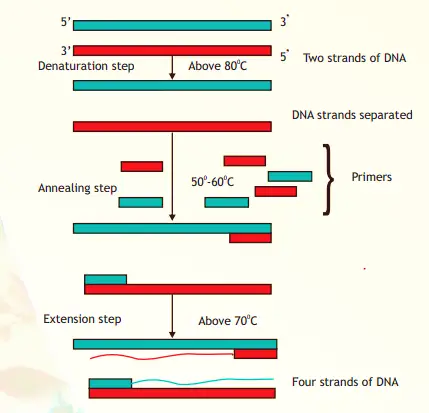
Working Mechanism of PCR:
The action of PCR includes several cycles. A single PCR amplification cycle involves these basic steps: Denaturation
The two strands of DNA are separated by applying a high temperature (95°C). After separation each strand acts as template for DNA synthesis. Primer Annealing
Each single strand anneals with a primer at a lower temperature between 50° - 60° inn such a way that extension can occur from it in a 5’-3’ direction. The annealing temperature (in °C) can be calculated using the formula- T= 2(AT) + 4(G+C). Extension (Polynomerisation)
In this step, the enzyme Taq Polymerase extends each primer using dNTPs and the DNA strand as template. The temperature for extension is around 70°C.
The procedure is repeated and each set of steps is considered as one cycle (i.e. denaturation, annealing and extension). At the end of one cycle two DNA molecules becomes four and this geometric progression occurs with each cycle.
Always after n number of cycles, 2n molecules of DNA are generated using single stranded DNA as template. Applications of PCR
The invention of PCR technique has revolutionized every aspect of modern biology.
To detect pathogens, microbiologists in the past used techniques based on culturing and detecting antibodies against enzymes or protein specific to the pathogen. They are grown slowly so their cells are found less in number in the infected cell/tissue. It is difficult to culture them on artificial medium. Apart from taking time, many of these procedures were not specific. Hence, for their diagnosis PCR-based assays have been developed. These detect the presence of certain specific sequences of the pathogen present in the infected cells/tissues.
PCR diagnosis is faster, safer and more specific because it does not use live pathogen.
PCR is also valuable tool in forensic science as large amounts of DNA can be amplified from the small amounts present at the crime site, for DNA fingerprinting analysis.
In recent years, PCR has also found use in detecting specific microorganisms from environmental samples of soil, sediments and water.
In humans, there are thousands of generic diseases. Mutations are also related to genetic diseases. Presence of faulty DNA sequence can be detected before establishment of diseases.
It is useful in parental diagnosis of genetic diseases.
Dna Probe
DNA probe is mainly used in searching the functional target DNA.
A DNA probe is a small fragment of DNA (small single stranded sequence of DNA) that recognize and bind to complementary sequence.
A DNA probe which is single stranded will bind to a complimentary sequence with the same base pairing rules (A-T and C-G and vice versa) and if the probe can be tagged with a fluorescent or radioactive label, the complimentary sequence can be located either in a cell nucleus or a gel chromatogram.
Hybridisation Techniques
Southern Hybridisation Technique
This technique of identifying and locating specific sequences in DNA gels using probes was invented in 1975 by Edward Southern and is named Southern Hybridization Technique. Principle:
A specific DNA fragment can be separated and identified in a heterologous population of DNA molecules on the basis of binding of DNA probe with its complementary DNA strand. Procedure:
The procedure involves isolation and digestion if total genomic DNA with one or more restriction enzymes. The DNA fragments thus generated are separated in agarose gels using the technique of electrophoresis.
Different DNA bands are formed on agarose gel which represents DNA fragments of varying sizes.
These fragments are transferred from gel to nylon or nitrocellulose membrane. The process of DNA transfer is called “blotting”.
In the blotting procedure the DNA fragment are forced from the gel onto the membrane by capillary action.
The membrane is baked briefly to fix DNA fragments to the membrane so that they do not diffuse during the next step of hybridization. The membrane will have the same pattern of DNA bands as the original gel.
The membrane is then treated with the single stranded labeled probe for an appropriate period after which the membrane is washed and either photographed under UV light (if probe labeled with fluorescent) or overlaid with a photographic film (if probe is radioactive).
The location of the probe is determined leading to the identification of a specific DNA fragment obtained from that given genomic DNA.
Dna Library
A gene library is the collection of different DNA sequences of an organism where each sequence has been cloned with a vector for ease of purification, storage and analysis.
A DNA library is not only containing all possible fragments of DNA from a given cell or organisms but also large amounts of the same as resources.
There are two types of libraries on the basis of source of DNA used:
Genomic Library (where genomic DNA is used)
cDNA Library (where cDNA or complementary DNA is produced using mRNA).
Genomic Library
It is the collection of clones that represent the complete genome of an organism. All fragments of DNA are inserted into vectors for further propagation into suitable host represent the entire genome of an organism.
To present a genomic library, the total genomic DNA is isolated from a tissue or organism and then fragmented using a restriction enzyme such as ECOR I.
An appropriate vector such as pBR322 (plasmid based) is also digested with the same enzyme and is then treated with the enzyme alkaline phosphatase which removes the 5’phosphate group to prevent the plasmid from self-ligation.
The DNA fragments and the cut vector are mixed and then treated with the enzyme DNA ligase.
Each vector molecule contains a different fragment of DNA and these are introduced into E.coli host cells by a technique called transformation.
A genomic DNA library has all possible DNA sequences in large amounts from the given cell type.
Genomic DNA library is larger than cDNA library.
cDNA Library
The library made from complementary or copy DNA (cDNA) is called cDNA library.
This library has two major advantages over a genomic library. Firstly, it represents only those genes that are being expressed by a particular cell or under specific conditions. Eukaryotes which include multicellular organisms such as animals and plants have differentiated cells e.g. liver and brain cells express some common proteins and also proteins which are not common.
Secondly, since the source material in constructing such libraries is mRNA, these molecules lack introns and hence would represent only the coding sequences of genome.
The cDNA library can be constructed by using mRNA and mRNAs are the highly processed, intron-free representatives of DNa having only coding sequences.
The construction of cDNA library begins with the isolation of mRNa from a given cell type or tissue which are copied into cDNA using a special enzyme called reverse transcriptase. The procedure results in double stranded cDNA which can be incorporated into vectors such as pBR322.

")